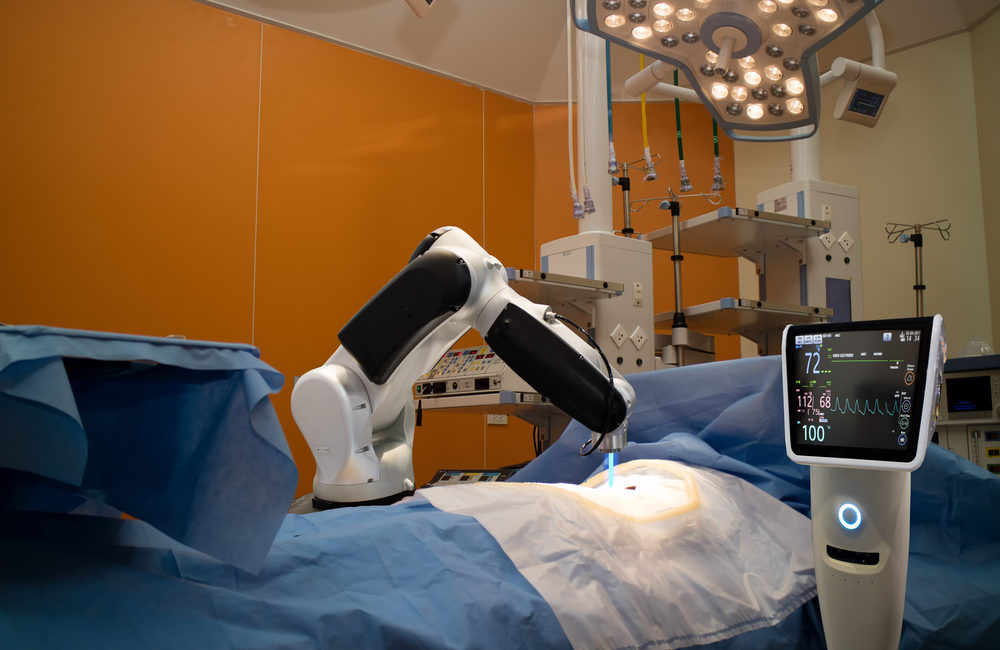
The U.S. Food and Drug Administration (FDA) has announced that Zimmer Biomet is recalling ROSA Brain 3.0 surgical robots due to a possible software defect. According to the recall announcement, there is a software problem that could cause the robotic arm to position itself incorrectly.
The incorrect positioning of the robotic arm could pose a risk to patients undergoing surgery. Due to this risk, the FDA has classified the recall as Class I, which is the most serious type of recall. A Class I recall means that using the device may cause “serious injuries or death.”
What is the ROSA Brain Surgical Robot?
The ROSA Brain surgical robot assists neurosurgeons during surgery. The robotic device helps surgeons place medical instruments or implants during brain surgery. The device includes a robotic arm that is attached to a touch screen. Different attachments can be added to the arm to help surgeons based on the procedure they are performing.
The ROSA Brain surgical robot acts similar to a GPS device, but instead of a road map, the device creates a map of the patient’s brain. The ROSA robot is used for various types of neurosurgery including:
- Placing electrodes to pinpoint seizure activity
- Using electrodes to treat epilepsy
- Performing biopsies of the brain
- Laser probing deep in the brain (laser ablation)
- Treating tumors with lasers
- Removing tumors in the brain
Robot-assisted brain surgery is considered less invasive and safer than traditional surgery. The mapping and visual assistance helps surgeons be more precise and lowers the risk of infection. There are around 20 hospitals in the U.S. who have ROSA Brain surgical robots. Because of the complex nature of surgical robots, surgeons must obtain special education in their proper use.
Details about the ROSA Brain Recall
Zimmer Biomet is recalling the ROSA Brain robotic device after discovering a software problem in versions V3.0.0.0 (v3.0.0.16) and v3.0.0.5 (v3.0.0.20). The software problem can result in the robotic arm driving to an incorrect position. The software problem creates a discrepancy between the initial skin markings and the trajectory of the instrument. This discrepancy poses a risk to the patient if not immediately corrected. So far, there are five complaints due to the software problem. Also, one patient reports injuries due to the device.
There are a total of 86 products in the recall. The list of product codes are included in the FDA recall announcement. All of the products in the recall were manufactured between February 2016 and December 2018. Distribution dates are between April 2016 and March 2019.
What to Do About the ROSA Brain Recall
The FDA is warning healthcare providers, neurosurgeons and patients to be aware of the possible danger. To reduce the risk of further injuries, Zimmer Biomet and the FDA offer the following:
- An Urgent Device Correction letter provides instructions for a “workaround” to resolve the issue.
- Zimmer Biomet is providing a label to surgical centers to adhere to the device alerting users of the risk and how to resolve the problem.
- Zimmer Biomet engineers are visiting customer sites to implement a new version of the software.
- Customers can expect additional information as updates are available.
- Zimmer Biomet lists contact, Perry Twyford, at 281-389-3236 for customer questions or concerns.
- Patients who experience an adverse event after brain surgery should report it to the FDA via the MedWatch Safety Information and Adverse Event Reporting Program.
Have Questions about a Medical Device Recall?
Medical device recalls can be scary, especially if you have recently had surgery or any type of medical procedure. If you have questions about a medical device recall, you can always contact Drug and Device Watch. Our team has extensive knowledge of medical devices, recalls and your legal rights as a patient.
To get answers to your questions, you can call us toll free at 1-888-458-6825. Or, you can fill out our online form to request more information. We are here to help!
Sources: